 |
Résumés des présentations | ||||
|
Résumés des présentations | ||||
|
2024, 17 déc. | F. Rebaudo | Modéliser la phénologie des insectes dans un contexte de réchauffement climatique | XXX |
|
||||
|
2024, 19 déc. | L. Camus | SOUTENANCE DE THÈSE Prédiction génomique du potentiel adaptatif de populations dans un nouvel environnement : application aux espèces envahissantes |
218 |
|
||||
|
2024, 19 nov. | J.F. Dufayard, L. Percevault & C. Blitz Frayet |
Où en est-on sur les données de la recherche ? | 217 |
|
||||
|
2024, 21 oct. | A. Cruaud | SOUTENANCE DE HDR Systématique intégrative des Chalcidiens (Hyménoptères) et régulation naturelle dans les agro-écosystèmes |
216 |
|
||||
|
2024, 15 oct. | L. Percevault | Partager des données interopérables en recherche : Focus sur les standards et les entrepôts | 215 |
|
||||
|
2024, 1er oct. | C. Roemer | Étude des communautés de Chiroptères par la méthode acoustique et applications possibles pour l'étude des interactions avec leurs proies | 214 |
|
||||
|
2024, 30 jul. | S. Hansen | Biological control of an invasive, highly damaging ambrosia beetle: the polyphagous shot-hole borer in South Africa | 213 |

|
||||
|
2024, 28 mai | A. Weyrich | Epigenetic signatures of social status in free-ranging spotted hyenas (Crocuta crocuta) | 212 |

|
||||
|
2024, 14 mai | C. Marino | Consequences of biological invasions on a global scale: addressing the multiple facets of threatened terrestrial diversity | 211 |
|
||||
|
2024, 23 avr. | A. Charalabidis | Effet des interactions inter- et intraspécifique et de la variabilité intraspécifique sur le comportement alimentaire de coléoptères carabiques | 210 |
|
||||
|
2024, 9 avr. | L. Borges Silva | Azores Islands as a Model System: Understanding the Biodiversity, Ecological and Genetic Patterns, along Gradients of Anthropogenic Disturbance | 209 |

|
||||
|
2024, 8 avr. | R. Leblois | SOUTENANCE DE HDR Inférence en génétique spatiale des populations vue par le prisme de la coalescence |
208 |
|
||||
|
2024, 29 mar. | L. Castañeda | Physiological adaptation to environmental stress in insects | 207 |
|
||||
|
2024, 26 mar. | L. Marescot | Forecasting the risk of locust outbreak | 206 |
|
||||
|
2024, 12 mar. | D. Bourguet | Présentation des Registered Reports (RR) et de PCI RR | 205 |
|
||||
|
2024, 29 fév. | E. Shimbori | Bridging gaps in the study of parasitoid wasps: a perspective from the global south | 204 |
|
||||
|
2024, 27 fév. | C. Fournier & P. Aventurier | Publier en Open Access et payer des APC (ou pas !) | 203 |

|
||||
|
2024, 6 fév. | L. Genty | Functional diversity and potential of weeds as forage and floral resources: the case of Mediterranean vineyards and olive groves |
202 |
|
||||
|
2024, 30 jan. | E. Charbonnel | SOUTENANCE DE THÈSE Informer sur l’origine géographique et le statut spécifique d’un ravageur invasif et cryptique : validation d’une méthodologie haut-débit pour le suivi de la mouche orientale des fruits, Bactrocera dorsalis |
201 |
|
||||
|
2024, 23 jan. | P. Nunes | Modelling the dispersal of invasive species using landscape variables | 200 |
|
||||
|
2024, 9 jan. | J. Cornuault | Empirical and methodological research in biogeography, population genetics and phylogenetics | 199 |
|
||||
|
2023, 8 déc. | E. Frago | SOUTENANCE HDR A multitrophic approach to the dynamics of insect herbivores: Experiments with plants, predators and insect symbionts |
198 |
|
||||
|
2023, 5 déc. | O. Grente | Better understanding the links between wolf depredation behavior, culling and depredation levels with an individual based model | 197 |
|
||||
|
2023, 5 déc. | M. Sidous | Using camera traps to assess the impact of land use on scavenger communities | 196 |
|
||||
|
2023, 21 nov. | A. Patenkovic | Honey bee of Serbia. A population-genetic perspective | 195 |
|
||||
|
2023, 21 nov. | M. Savic Veselinovic | Response to heavy metal stress in Drosophila species. The metallothionen genes expression | 194 |
|
||||
|
2023, 17 nov. | T. Tsuchida | Investigating the mysteries of gall formation: A study of gall-forming weevil and its symbiotic bacteria | 193 |

|
||||
|
2023, 7 nov. | N. Gompel | Diversification of pigmentation patterns among Drosophila species | 192 |

|
||||
|
2023, 31 oct. | M. Bouilloud | Soutenance de thÈse Impacts de l'anthropisation sur les liens entre les communautés de petits mammifères, leur microbiote et les dangers zoonotiques |
191 |
|
||||
|
2023, 27 oct. | D. Teulon | The fall armyworm invasion of New Zealand | 190 |
|
||||
|
2023, 27 oct. | A. McGaughran | Adaptive processes through time in a global agricultural pest moth | 189 |
|
||||
|
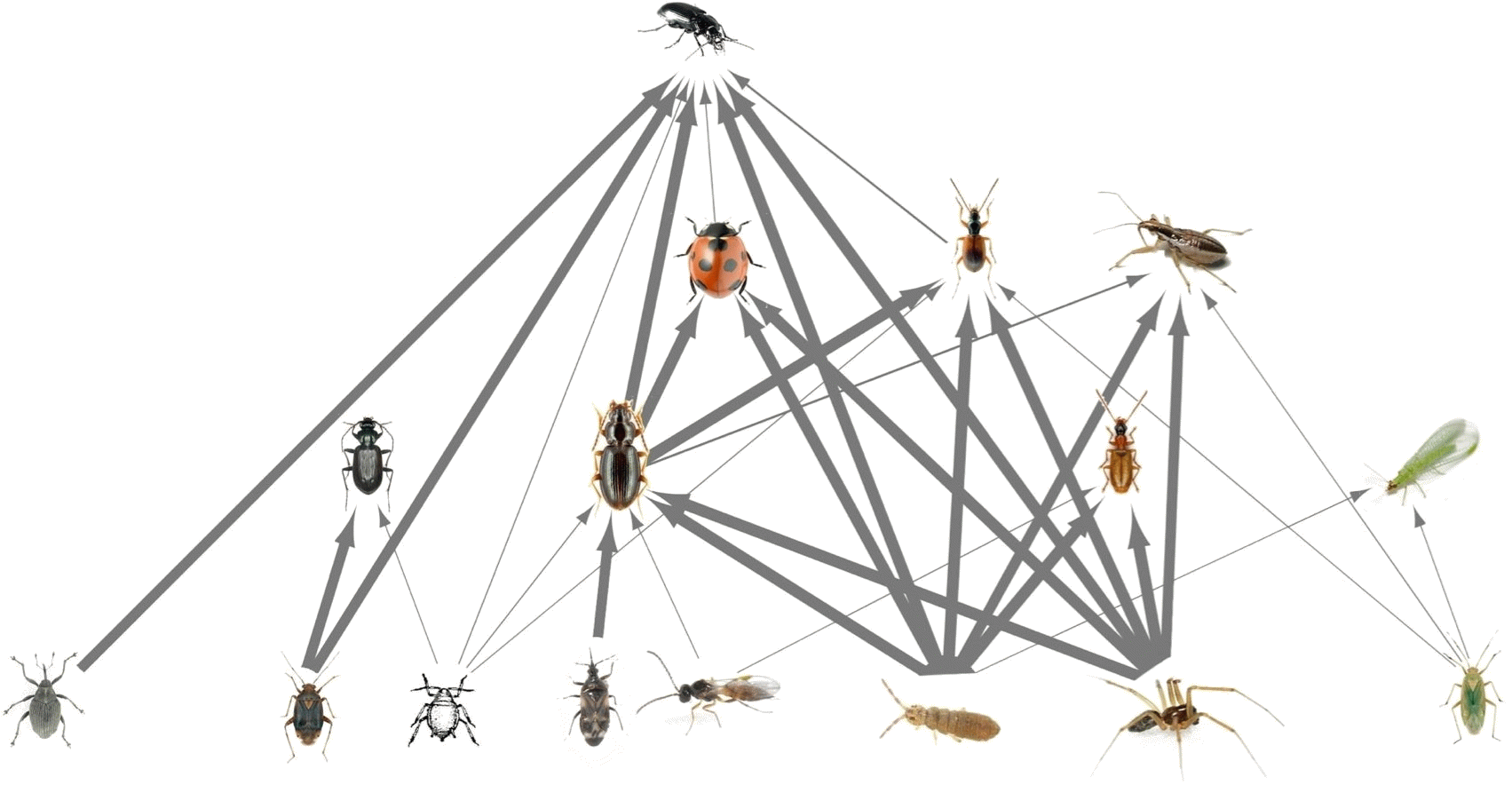
2023, 3 oct. | D. Bohan | Exploring the web of dark interactions | 188 |
|
||||
|
2023, 18 sep. | T. do Prado | Tiny Titans: Exploring the Potential of Soil Mites in Sustainable Agriculture | 187 |
|
||||
|
2023, 24 aoû. | M. Stastny | Regional adaptation of Integrated Pest Management to control invasive insects | 186 |
|
||||
|
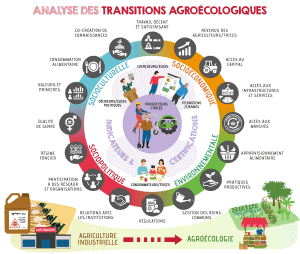
2023, 20 juin | A. Sosa | Pluridimensionnalité et participation dans l'évaluation des transitions agroécologiques : aspects environnementaux, socioéconomiques, socioculturels et sociopolitiques |
185 |
|
||||
|
2023, 15 juin | I. Poinas | Soutenance de thÈse : Impact des pratiques agricoles sur la dynamique spatio-temporelle des communautés de plantes et de coléoptères des bords de champs : approches fonctionnelles et multi-échelles |
184 |
|
||||
|
2023, 9 mai | A. Fraimout | Repeated evolution during habitat transitions from marine to freshwater | 183 |
|
||||
|
2023, 18 avr. | C. Pelosi | Soil engineers and their tremendous activity in a changing environment | 182 |
|
||||
|
2023, 4 avr. | A. Milcu | Ecotrons, powerful and versatile ecosystem analysers for ecology, agronomy and environmental sciences | 181 |
|
||||
|
2023, 21 mar. | M.C. Bopp | 40-year changes in weed communities induced by management and climate change in Mediterranean vineyards | 180 |
|
||||
|
2023, 14 mar. | V. Tiersen | Présentation de la société Green Score Capital, consultations en impact environnemental | 179 |
|
|
||||
|
2023, 14 mar. | V. Tiersen & M. Faucheux | Measuring the Environmental Impact of Industries FootPrint Target Tool Focus on assessing habitat degradation and structural connectivity |
179 |
|
||||
|
2023, 7 mar. | D. Percy 1 D. Burckhardt 2 |
1. Using molecular systematics to investigate patterns of diversity and speciation in psyllids 2. Psyllid pests and taxonomy |
178 |
|
||||
|
2023, 14 févr. | C. Vernier | SOUTENANCE DE THÈSE Mouvements collectifs et plasticité phénotypique : étude du polyphénisme de phase des locustes à plusieurs échelles spatio-temporelles |
177 |
|
||||
|
2023, 8 févr. | B. Guinet | Domestication virale chez les insectes parasitoïdes : une approche globale | 176 |
|
||||
|
2023, 8 févr. | J. Rouil | SOUTENANCE DE THÈSE Évolution et dynamique du système di-symbiotique chez les pucerons du genre Cinara |
175 |
|
||||
|
2023, 7 fév. | Y. Outreman | Les microbes prennent-ils le pouvoir ? | XXX |
|
||||
|
2023, 24 jan. | G. Dobigny | SÉMINAIRE INTERNE Écologie de la santé et suivi des micromammifères urbains à Antananarivo : protocoles et premiers résultats |
174 |
|
||||
|
2022, 21 nov. | I. Morianou | Understanding the resistance landscape to gene drive | 173 |
|
||||
|
2022, 18 octobre | R. Villoutreix | Ecological discontinuity packages genes into discrete units of diversity | 172 |
|
||||
|
2022, 5 juillet | M.S. Tixier, C. Cornier & A. François | Le département Biologie & Écologie de l'Institut Agro Montpellier : organisation, formations et partenariats | 171 |
|
||||
|
2022, 4 juillet | T. Strive | 70 years of Biological Control of Rabbits in Australia - An Ongoing Co-Evolutionary Arms-Race | 170 |
|
||||
|
2022, 28 juin | N. Guilpart | Europe's soybean self-sufficiency under climate-change: insights from data-driven yield projections using machine-learning | 169 |
|
||||
|
2022, 19 mai | M. Javal | Surveillance d'un cas extrême de changement d'hôte : vers une compréhension de la biologie, de l'écologie et du potentiel d'invasion d'un ravageur émergent de la canne à sucre en Afrique | 168 |
|
||||
|
2022, 12 mai | J. Kincaid-Smith | Schistosomiasis, hybridization and zoonosis: towards an integrative view of the pathosystem | 167 |
|
||||
|
2022, 5 mai | S. Serga | Wolbachia in natural populations of Drosophila from Ukraine – eighteen years of investigations | 166 |
|
||||
|
2022, 26 avr. | J. Gauthier | La muséomique: une fenêtre sur l’histoire passée des populations | 165 |
|
||||
|
2022, 19 avr. | E. Call | The age of museomics: how to get genomic information from museum specimens of Lepidoptera | 164 |

|
||||
|
2022, 15 avr. | C. Piou | SOUTENANCE HDR La modélisation écologique pour la gestion des populations de locustes |
163 |
|
||||
|
2022, 24 mar. | L. Henckel | Concilier production et conservation dans les écosystèmes fortement gérés (milieux agricoles et forêts productives) | 162 |
|
||||
|
2022, 22 mar. | P. Tresson | Détection et classification d'animaux sur des séquences d'images | 161 |
|
||||
|
2022, 8 mar. | C. Lutrat | Développement de souches de sexage génétique pour la Technique de l’Insecte Stérile chez les moustiques du genre Aedes | 160 |
|
||||
|
2022, 15 fév. | D. Santos García | A song of sap and blood | 159 |
|
||||
|
2022, 6 jan. | S. Fellous | Présentation du réseau ENI-BC+ : étude des Effets Non-Intentionnels des pratiques et techniques de protection des plantes, des animaux et gestion des vecteurs. | 158 |
|
||||
|
2021, 14 déc. | G. Krouk | Some examples of how we use machine learning in plant science and beyond | 157 |
|
||||
|
2021, 30 nov. | R. Allio | Phylogenomics and comparative genomics: the case of coevolution between the swallowtail butterflies (Papilionidae) and their host plants | 156 |
|
||||
|
2021, 19 oct. | M. Rousselle | Population size, incomplete lineage sorting and selection in animal genomes | 155 |
|
||||
|
2021, 1er jun. | J.F. Martin | Journaux prédateurs, où est la limite? Une discussion autour des raisons pour éviter les journaux douteux et tenter de les détecter |
154 |
|
||||
|
2020, 13 avr. | B. Michel | Éléments de Neuroptérologie africaine : fourmilions et ascalaphes | 153 |
|
||||
|
2021, 25 mar. | J.F. Martin | La gestion des données de recherche – Devons-nous vraiment tout changer ? | 152 |
|
||||
|
2021, 16 mar. | G. Castel | Diversité et évolution de l'Orthohantavirus Puumala en France | 151 |
|
||||
|
2021, 2 mars | Q. Rougemont | Demographic history shaped within species variation in the deleterious mutation load in a broadly distributed Pacific Salmon | 150 |
|
||||
|
2021, 9 fév. | G. Delvare | Les Chalcidiens (Hymenoptera, Chalcidoidea) des asphodèles | 149 |
|
||||
|
2021, 2 fév. | F. Mallard | The architecture of genetic adaptation during experimental evolution in a novel thermal environment | 148 |
|
||||
|
2021, 26 jan. | A. Mercier & L. Galal | Introgressions massives de souches du parasite Toxoplasma gondii de l'ancien Monde vers le nouveau Monde : un jeu du chat et de la souris de part et d'autre de l'Atlantique | 147 |

|
||||
|
2020, 4 déc. | S. Madrières | SOUTENANCE THÈSE Étude des interactions entre l'orthohantavirus Puumala et son réservoir dans l'épidémiologie de la néphropathie épidémique |
146 |
|
||||
|
2020, 6 oct. | M. Bogaerts Marquez | Insights into the environmental pressures driving adaptation in Drosophila melanogaster | 145 |
|
||||
|
2020, 25 sep. | E. Frago | A meta-analysis on the benefits and costs of hosting secondary endosymbionts in sap-sucking insects | 144 |
|
||||
|
2020, 15 sep. | R. Guilhot | Exposé soutenance thèse Écologie des drosophiles et de leurs symbiotes microbiens en conditions naturelles |
143 |
|
||||
|
2020, 25 jun. | C. Filippone | Investigation of emerging zoonotic viruses in Europe and Africa | 142 |
|
||||
|
2020, 23 jun. | C. Perrier | Local adaptation in blue tit populations: insights from population genomics | 141 |
|
||||
|
2020, 8 jun. | M. Ollivier | SOUTENANCE THÈSE Lutte biologique par introduction contre Sonchus oleraceus (Asteraceae) |
140 |
|
||||
|
2020, 29 mai | C. Meynard | SOUTENANCE HDR Aires de répartitions, métacommunautés et biodiversité: du théorique à l’appliqué |
139 |
|
||||
|
2020, 5 mai | N. Rode | Multiple hybrid origins of invasive diploid and polyploid asexual lineages of the brine shrimp Artemia | 138 |
|
||||
|
2020, 10 mar. | M. Arias | Effect of range shifts in trophic interactions of two harmful moths in a global change context | 137 |
|
||||
|
2020, 25 fév. | C. Favret | Les 5 ‘D’s de la taxonomie : un guide d’utilisateur | 136 |

|
||||
|
2019, 17 déc. | M.S. Tixier | Les acariens Phytoseiidae, de la taxonomie à la lutte biologique | 135 |
|
||||
|
2019, 13 déc. | L. Olazcuaga | SOUTENANCE THÈSE Réponses adaptatives chez Drosophila suzukii, une espèce généraliste envahissante |
134 |
|
||||
|
2019, 13 déc. | O. Tournayre | SOUTENANCE THÈSE Structure et fonctionnement des colonies de Grand rhinolophe dans l'Ouest de la France |
133 |
|
||||
|
2019, 3 déc. | F. Ecke | Ecology of hantavirus infections in Sweden: Drivers of the contact zone between the virus, rodents and humans | 132 |
|
||||
|
2019, 12 nov. | N.T. Dianzinga | Can dispersal explain the coexistence of two predators in reciprocal intraguild predation? | 131 |

|
||||
|
2019, 15 oct. | E. Jousselin | Évolution des systèmes di-symbiotiques chez les pucerons | 130 |
|
||||
|
2019, 1er oct. | C. Brouat | Parasitisme et invasions biologiques : le cas du rat noir et de la souris domestique au Sénégal (2012-2019) | 129 |
|
||||
|
2019, 26 sep. | D. Navia | Metabarcoding applied to biological control involving microarthropods: early steps and prospects | 128 |
|
||||
|
2019, 24 sep. | V. Baudrot | Tracking uncertainties in mechanistic models for risk assessment of pest control strategies | 127 |
|
||||
|
2019, 17 sep. | A. Sheppard | Bilan des stratégies et des opportunités de recherche du CSIRO | 126 |
|
||||
|
2019, 10 sep. | J.E. Cohen | Vector-borne disease control and Taylor's law of fluctuation scaling: Chagas disease in Argentina | 125 |
|
||||
|
2019, 3 sep. | M.D. Greenfield | Animal choruses emerge from receiver psychology | 124 |
|
||||
|
2019, 23 jul. | S. Catalano | Human schistosomiasis in the Senegal river basin: does wildlife matter? | 123 |
|
||||
|
2019, 2 jul. | E. Vercken | Vagues tirées, vagues poussées : causes et conséquences en écologie | 122 |
|
||||
|
2019, 3-4 jun. | Le printemps de Baillarguet 2019 : les journées des non-titulaires du campus | 121 |
|

|
||||
|
2019, 21 mai | L. Granjon | Retours d'information vers les populations sur l'expansion de rongeurs invasifs au nord Sénégal | 120 |
|
||||
|
2019, 16 avr. | R. Streiff | Spécialisation d'un insecte ravageur des cultures et approche de biologie évolutive pour la gestion des populations : le point des recherches sur les pyrales du genre Ostrinia | 119 |
|
||||
|
2019, 12 avr. | A. Rombaut | Soutenance thèse Comprendre et lutter contre les dégâts du ravageur Drosophila suzukii sur l'agro-système vigne |
118 |
|
||||
|
2019, 2 avr. | E. Frago | Symbionts protect aphids from parasitic wasps by attenuating herbivore-induced plant volatiles | 117 |
|
||||
|
2019, 26 mar. | G. Fried | Exposé soutenance HDR Apports des approches fonctionnelles pour l'évaluation des risques associés aux changements de végétation induits par les activités humaines |
116 |
|
||||
|
2019, 19 mar. | M. Barberà | Circadian clock and photoperiodism in the pea aphid Acyrthosiphon pisum | 115 |
|
||||
|
2019, 12 mar. | R. Vitalis | Inférence de l'histoire évolutive des populations | 114 |
|
||||
|
2019, 19 fév. | V. Pavinato | Joint inference of demography and selection from genomic temporal data using approximate Bayesian computation | 113 |
|
||||
|
2019, 5 fév. | J.Y. Rasplus, J.P. Rossi, M. Chartois & A. Cruaud | Caractériser les réseaux d’interactions plantes-vecteurs pour mieux gérer et anticiper la progression du phytopathogène Xylella fastidiosa | 112 |
|
||||
|
2019, 22 jan. | G. Kergoat | Étude des trajectoires évolutives de communautés d'insectes au moyen d'approches macroévolutives | 111 |
|
||||
|
2019, 8 jan. | N. Charbonnel | Le risque d'émergence de maladies zoonotiques à travers le prisme de l'immunoécologie et de l'immunogénétique : les rongeurs et hantavirus responsables de fièvres hémorragiques | 110 |
|
||||
|
2018, 14 déc. | V. Hivert | SOUTENANCE THÈSE Analyse de la différenciation génétique à l'ère des nouvelles technologies de séquençage |
109 |
|
||||
|
2018, 11 déc. | A. Estoup | Phéno-génomique des populations d'espèces envahissantes : études en cours chez Harmonia axyridis et Drosophila suzukii | 108 |
|
||||
|
2018, 20 nov. | F. Vanlerberghe | La spécialisation à la plante-hôte chez le puceron Aphis gossypii : comment ça marche ? | 107 |
|
||||
|
2018, 9 nov. | S. Boitard | Estimation de l'histoire démographique d'une population à partir de données génomiques haut-débit | 106 |
|
||||
|
2018, 6 nov. | C. Kerdelhué | Différenciation allochronique chez la processionnaire du pin Thaumetopoea pityocampa : passé, présent et futur d'une population atypique | 105 |
|
||||
|
2018, 9 oct. | L. Ballesteros | Saturniid and sphingid moths as novel models for the study of insect diversity and macroecology | 104 |
|
||||
|
2018, 25 sep. | S. Blanchet | Changements globaux et écosystèmes aquatiques : une opportunité pour combiner recherche fondamentale et appliquée | 103 |
|
||||
|
2018, 11 sep. | L. Borner | Modéliser la répartition de ravageurs des cultures : défis méthodologiques et projections climatiques | 102 |
|
||||
|
2018, 3 jul. | D. Bourguet | Peer Community in: un système de recommandation public et gratuit de preprints | 101 |
|
||||
|
2018, 26 jun. | K. Maeno | Propositions to improve the preventative management of Desert locusts based on new knowledge of its ecology | 100 |
|
||||
|
2018, 5 jun. | I. Bravo | Asymptomatic, acute or chronic infections: a twisted road from genotype to phenotype in Papillomaviruses | 99 |
|
||||
|
2018, 28-29 mai | Le printemps de Baillarguet 2018 : les journées des non-titulaires du campus | 98 |
|
|
||||
|
2018, 10 avr. | U. Berger | Is our world agent-based and can we model it? | 97 |
|
||||
|
2018, 3 avr. | V. Caron | Attack of the clones: the introduced willow sawfly Nematus oligospilus in Australia | 96 |
|
||||
|
2018, 20 mar. | V. Pavinato | Influence of historical land use and modern agricultural expansion on the spatial and ecological divergence of sugarcane borer, Diatraea saccharalis (Lepidoptera: Crambidae) in Brazil | 95 |
|
||||
|
2018, 14 mar. | S. Madrières | Dynamique et évolution intra-hôte du virus Puumala dans des zones géographiques épidémiologiquement différentes | 94 |
|
||||
|
2018, 13 fév. | A. Sow | Régulation naturelle des populations de la mineuse de la chandelle de mil, Heliocheilus albipunctella (Lepidoptera, Noctuidae) | 93 |
|
||||
|
2018, 6 fév. | J. McCutcheon | Genome fragmentation in endosymbionts: good, bad or just ugly? | 92 |

|
||||
|
2017, 19 déc. | M. Godefroid | Combining genetic and distributional data for a better understanding of the drivers that shape the distribution of plant pests | 91 |
|
||||
|
2017, 6 déc. | S. Fellous | The ecology of host-microbe interactions: from the study of model organisms to the protection of crops against pest arthropods | 90
|
|
||||
|
2017, 6 déc. | P. Becher | Interactions between the yeast Hanseniaspora uvarum and the spotted wing Drosophila, Drosophila suzukii | 89
|
|
||||
|
2017, 21 nov. | E. Saulnier | EBOV phylodynamics using regression-ABC | 88 |
|
||||
|
2017, 7 nov. | S. Ramos-Onsins | Bioinformatic and analytical tools for the analysis of whole-genome sequence polymorphism data | 87 |
|
||||
|
2017, 26 sep. | R. Garnier | Evolutionary ecology of the dynamics of antibodies | 86 |
|
||||
|
2017, 19 sep. | A. Fraimout | Évolution de la variation génétique et phénotypique au cours d'une invasion : le cas de Drosophila suzukii | 85 |
|
||||
|
2017, 30 jun. | E. Petit | Drivers of the dynamics and genetics of lesser horseshoe bat populations | 84 |

|
||||
|
2017, 20 jun. | N. Rode | Fitness effect of mutations between phases of the life cycle of the root-rot fungus Heterobasidion parviporum | 83 |
|
||||
|
2017, 6 jun. | J. Collet | Pleiotropy: its extent, modular organisation and how selection operates on it. Lessons from gene expression of Drosophila serrata | 82 |
|
||||
|
2017, 30 mai | H. Svardal | Locating introgression and non-tree-like ancestry on a large phylogeny | 81 |
|
||||
|
2017, 23-24 mai | Le printemps de Baillarguet 2017 : les journées des non-titulaires du campus | 80 |
|

|
||||
|
2017, 16 mai | J. Réveillaud | Étude du rôle du microbiote à partir de données métagénomiques shotgun | 79 |
|
||||
|
2017, 11 mai | Journée thématique : Réseaux, invasions et émergences | 78 |
|
|
||||
|
2017, 2 mai | E. Lievens | Ecological and evolutionary factors drive the specialization of two microsporidian parasites of native and invasive Artemia | 77 |
|
||||
|
2017, 27 avr. | F. Moreau | Produits & services proposés par l'Agence d'information génétique ADNid | 76 |

|
||||
|
2017, 21 mar. | A. Manzano | Evolution of co-obligate symbioses in aphids and genome reduction in endosymbionts | 75 |
|
||||
|
2017, 16 mar. | L. Brousseau | Génomique de la spécialisation à la plante hôte chez deux espèces sœurs de pyrales (Ostrinia nubilalis et O. scapulalis) | 74 |
|
||||
|
2017, 21 fév. | D. Abu-Awad | The consequences of demographic stochasticity on fixation | 73 |
|
||||
|
2017, 14 fév. | F. Riquet | Du design aux analyses bio-informatiques : de nouveaux marqueurs pour une espèce protégée, l’hippocampe moucheté Hippocampus guttulatus | 72 |
|
||||
|
2017, 31 jan. | R. Hufbauer | Is evolution a driver or passenger of biological invasions? Empirical data from experimentally invading Tribolium populations, and open questions for how to proceed with possible genomic analyses | 71 |
|
||||
|
2017, 24 jan. | A. Gilabert | Emergence de pathogènes : évolution du parasitisme et adaptation à l'hôte | 70 |
|
||||
|
2017, 18 jan. | A. Sheppard | Classical biological control research at CSIRO: new directions for vertebrates, invertebrates and weeds | 69 |
|
||||
|
2016, 16 déc. | A. Dubois | Mieux comprendre la répartition de l'hantavirus Puumala au sein des populations de campagnols roussâtres en Europe : aspects immunologiques et génomiques | 68 |
|
||||
|
2016, 13 déc. | M. Leitwein | Approche génomique de l'impact des déversements de truites (Salmo trutta) domestiques dans les populations sauvages d'origine méditerranéenne. | 67 |
|
||||
|
2016, 29 nov. | P. Stuart | The bank vole invasion in Ireland as a model system to study parasite dynamics and immunogenetics of invaders and natives | 66 |
|
||||
|
2016, 22 nov. | C. Silvy | Nos publications 2013-2015 : indicateurs bibliométriques traditionnels et alternatifs. Débats sur l'évaluation de la recherche | 65 |
|
||||
|
2016, 15 nov. | R. Oliva Molina | Physical barriers vs pest. Laboratory for quality control and evaluation of agrotextiles | 64 |
|
||||
|
2016, 4 oct. | O. Bouchez | La plateforme Génome et Transcriptome de Toulouse | 63 |

|
||||
|
2016, 20 sep. | D. Carrasco | Do you remember when? Effect of past experiences in future decisions in a noctuid moth | 62 |
|
||||
|
2016, 5 jul. | J.C. Aymes | Retour d'expérience sur la gestion de l'activité vidéo dans un laboratoire de recherche en écologie comportementale des poissons diadromes | 61 |
|
||||
|
2016, 28 jun. | S. de Mita | Conséquences génétiques d’un épisode d’adaptation à l'hôte chez l’agent de la rouille foliaire du peuplier | 60 |
|
||||
|
2016, 23 jun. | C. Guédot | The establishment of a new pest: phenology, crop susceptibility and impact of landscape on Drosophila suzukii | 59 |
|
||||
|
2016, 21 jun. | B. Gourbal et G. Mitta | Un parasitoïde utilise un virus pour modifier le comportement de son hôte | 57 |
|
||||
|
2016, 7 jun. | E. Artige | De la collecte aux vouchers moléculaires : la gestion des collections au CBGP | 56 |
|
||||
|
2016, 2-3 jun. | Le printemps de Baillarguet 2016 : les journées des non-titulaires du campus | 55 |
|

|
||||
|
2016, 19 mai | D. Waller | Causes and consequences of exotic invasions in Wisconsin forests | 54 |
|
||||
|
2016, 17 mai | M. Orsucci | Spécialisation à la plante hôte et isolement reproducteur chez des lépidoptères ravageurs de cultures | 53 |

|
||||
|
2016, 14 avr. | ABC-day and more at CBGP, Montpellier | 52 |
|
|
||||
|
2016, 7 avr. | F. Clemente | Kimtree: dealing with ascertainment bias and selection using SNP data | 51 |
|
||||
|
2016, 29 mar. | S. McCairns | Tipping the scales. Other lessons in adaptive evolution from the three-spine stickleback | 50 |
|
||||
|
2016, 15 mar. | A. Appelgren | Conséquences évolutives de la structuration des populations d'un ectoparasite à différentes échelles spatiales : approches empiriques sur le système puce des oiseaux-passereaux |
49 |
|
||||
|
2016, 8 mar. | F. Mahé | Vers un meilleur filtrage des données de séquençage pour les analyses de diversité environnementale | 48 |
|
||||
|
2016, 16 fév. | C. Walton | The evolution of malaria vectors in Southeast Asia | 47 |
|
||||
|
2016, 2 fév. | J. Claude | Variations du phénotype crânien dans les populations naturelles chez les vertébrés : perspectives à l'interface entre morphométrie géométrique, génomique et génétique quantitative | 46 |
|
||||
|
2015, 16 déc. | V. Thouzeau | Inférence statistique des changements culturels et démographiques en Asie Centrale à l'aide de données linguistiques et génétiques | 45 |
|
||||
|
2015, 11 déc. | C. Diagne | Communautés de parasites, immunité et succès d’invasion de rongeurs commensaux : cas du rat noir et de la souris domestique au Sénégal | 44 |
|
||||
|
2015, 1er déc. | E. Charles | Présentation de la société montpelliéraine ApoH-Technologies | 43 |
|
||||
|
2015, 17 nov. | T. Decaëns | Barcoding ADN et traits fonctionnels pour l'étude de la dynamique des communautés de vers de terre tropicaux | 42 |
|
||||
|
2015, 3 nov. | Y. Desdevises | Interactions coévolutives entre microalgues et virus géants | 41 |
|
||||
|
2015, 30 oct. | B. Linard | Metagenome skimming and large-scale comparative genomics of complex arthropod communities | 40 |
|
||||
|
2015, 26 oct. | M. Hoodle | An overview of the biocontrol program targeting Asian citrus psyllid in California | 39 |

|
||||
|
2015, 23 oct. | J. Pisano | SOUTENANCE THÈSE Histoires évolutives de rongeurs holarctiques : approche micro- et macroévolutive |
38 |
|
||||
|
2015, 1er oct. | N. Ali | Communautés de nématodes phytoparasites associées à l'olivier : réponses aux forçages anthropiques et environnementaux | 37 |
|
||||
|
2015, 29 sep. | G. Fried | L’apport des suivis à grande échelle pour la compréhension de la dynamique de la flore des champs cultivés | 36 |
|
||||
|
2015, 22 sep. | G. Thébaud | Modélisation spatio-temporelle de la dispersion d'un virus de plantes pour estimer des paramètres, hiérarchiser leur influence et optimiser la gestion de l'épidémie | 35 |
|
||||
|
2015, 4 sep. | E. Toussaint | Intégration des approches moléculaires pour l'étude de la systématique et des dynamiques macro-évolutives des insectes | 34 |
|
||||
|
2015, 15 jul. | J.C. Illera | Colonisation, diversification and extinction in Macaronesian birds: a synthesis of phylogenetic and fossil information | 33 |

|
||||
|
2015, 9 jul. | T. Brévault | Arrêt sur un programme de recherche à Biopass, Dakar, Sénégal. La biodiversité au service de la régulation des insectes ravageurs des cultures |
32 |
|
||||
|
2015, 9 jul. | A. Chailleux | Lutte biologique contre des espèces invasives : quelles sont les perspectives offertes par la mobilisation de plusieurs disciplines ? | 31 |
|
||||
|
2015, 7 jul. | D. Navia | Les ravageurs invasifs. Un défi pour l'agriculture au Brésil | 30 |
|
||||
|
2015, 30 jun. | M. Navascuès, A. Becheler et R. Vitalis | Tracking adaptation to environmental changes in experimental or monitored populations: evaluation of a method to detect loci under selection | 29 |
|
||||
|
2015, 16 jun. | J.M. Duplantier | L'invasion du rat noir (Rattus rattus) en Afrique de l'Ouest : modalités, chronologie et situation actuelle | 28 |

|
||||
|
2015, 4-5 jun. | Le printemps de Baillarguet 2015 : les journées des non-titulaires du campus | 27 |
|

|
||||
|
2015, 2 jun. | J. Abbate | Is recovery more important than resistance? Elevational disease distribution in a natural plant pathogen system: Insights from genetic variation in avoidance and recovery resistance. | 26 |
|
||||
|
2015, 19 mai | M. Joron | Natural selection and the formation of a mimicry supergene in butterflies | 25 |
|
||||
 Supergene alleles and mimicry polymorphism in Heliconius numata |
||||
|
2015, 5 mai | S. Jaulin et B. Louboutin | Présentation des activités 2013-2014 de l'antenne régionale de l'OPIE | 24 |
|
||||
|
2015, 28 avr. | C. Morris et M. Barbier (animateurs) | Mieux gérer les émergences de bioagresseurs Séminaire de discussion et débat autour de cas d’études pour renforcer les liens interdisciplinaires entre Sciences de la Vie et Sciences sociales |
23 |
|
||||
|
2015, 17 mar. | P. Cruaud | Sources hydrothermales et suintements froids : des écosystèmes marins profonds basés sur la chimiosynthèse microbienne | 22 |
|
||||
|
2015, 3 mar. | P. Reynaud, G. Fried et J.F. Germain | Hébergée au CBGP, une agence évalue les risques phytosanitaires vis-à-vis des arthropodes et des plantes invasives | 21 |
|
||||
|
2015, 17 fév. | J.C. Streito | Où en est le barcoding des arthropodes au CBGP ? Point d'étape et évolution vers les approches à haut débit. | 20 |
|
||||
|
2015, 3 fév. | L. Smith * | Current Research at the European Biological Control Laboratory | 19 |
|
||||
|
2015, 20 jan. | J. Le Fur | Modélisation multi-facteurs de la dynamique d'une population de campagnols des champs. Hiérarchisation des déterminants biologiques, environnementaux, sociaux. | 18 |
|
||||
|
2015, 6 jan. | C. Silvy | De l'Open Access à l'Open Data : enjeux et perspectives | 17 |
|
||||
|
2014, 16 déc. | Bertrand Gauffre1-2, S. Mallez2-3, M.P. Chapuis4, R. Leblois4, I. Litrico5, S. Delaunay5 & I. Badenhausser1-2 | Spatial heterogeneity in landscape structure influence dispersal and the genetic structure of populations: empirical evidence from a grasshopper in an agricultural landscape | 16 |
|
||||
|
2014, 4 déc. | Z. Boratynski | Adaptation of rodents to arid environments: Jaculus, colour and remote sensing data | 15 |
|
||||
|
2014, 2 déc. | S. Cornet | Favorisation de la transmission : manipulation parasitaire et plasticité chez Plasmodium. Le cas de la malaria aviaire. | 14 |
|
||||
|
2014, 18 nov. | R. Leblois | Inférences de changements passés de tailles de populations à partir de données microsatellites et de séquences d'ADN | 13 |
|
||||
|
2014, 8 jul. | B. Villegas Ramirez | A targeted enrichment strategy for the sequencing of medicinal species in the Indonesian Flora | 12 |
|
||||
|
2014, 17 jun. | C. Meynard | Climate change and biogeography: matching genetic data with past and future climate change using species distribution models | 11 |
|
||||
|
2014, 27 mai | J.E. Longueville | Quels gènes pour quelles plantes hôtes ? Une approche de génomique pour étudier l'adaptation à la plante hôte chez l'acarien Tetranychus urticae | 10 |
|
||||
|
2014, 29 avr. | J. Pisano | Out-of-Himalaya: evolutionary history of Dipodoidea (Rodentia) evidences the impact of past Asian environmental changes | 9 |
|
||||
|
2014, 22 avr. | D. Bourguet | Impacts sanitaires et environnements liés aux cultures de plantes transgéniques. Forces et faiblesses de l'évaluation des risques (2/2) | 8 |
|
||||
|
2014, 15 avr. | D. Bourguet | Impacts sanitaires et environnements liés aux cultures de plantes transgéniques. Forces et faiblesses de l'évaluation des risques (1/2) | 7 |
|
||||
|
2014, 1er avr. | C. Diagne | Immunité et succès d'invasion : premiers résultats sur la souris (Mus musculus domesticus) au Sénégal | 6 |
|
||||
|
2014, 18 mar. | A. Sanchez Meseguer | Spatio-temporal evolution of the genus Hypericum (Hypericaceae): the strategy of the marathon runner | 5 |
|
||||
|
2014, 4 mar. | F. Chardonnet * | Rôle du gène foraging dans l’évolution du comportement alimentaire de noctuelles foreuses de céréales | 4 |
|
||||
|
2014, 18 fév. | C. Bonneaud * | Immunoécologie des oiseaux | 3 |
|
||||
|
2014, 4 fév. | N. Tordo * | Exploration de la diversité génétique des hantavirus en France métropolitaine et ultramarine | 2 |
|
||||
|
2014, 23 jan. | C. Piou | Coupling historical prospection data and a remotely-sensed vegetation index for the preventative control of Desert locusts | 1 |
|
||||